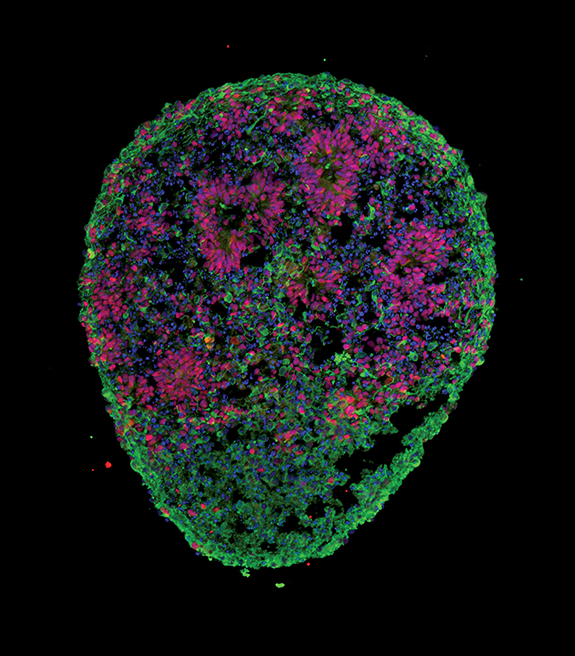
Brain 'organoids,' similar to the one shown here, are revealing how the neurons of individuals with autism spectrum disorders differ from those of unaffected family members. J. Mariani et al. Cell 162, 375-390 (2015).
In 2015, the Simons Foundation Autism Research Initiative (SFARI) supported nearly 200 autism researchers. These SFARI Investigators have pushed forward the frontiers of autism research in many ways, uncovering more and more of the condition's genetic architecture and also elucidating higher-level biological pathways, behavior and cognition. The following pages present some highlights of the past year's research by SFARI Investigators.
Brain 'organoids' grown from stem cells derived from skin cells of four boys with autism display shared biological anomalies, a study shows, even though none of the boys have any of the mutations that have so far been associated with autism risk. The research, published in the July 16, 2015 issue of Cell, supports the idea that the heterogeneous genetic underpinnings of autism converge on a much smaller set of biological pathways.
The researchers, led by SFARI Investigator Flora Vaccarino of Yale University, grew organoids — three-dimensional neural aggregates that mimic some features of the developing brain — from cells of four boys with unusually large heads, a feature that appears in about 20 percent of individuals with autism. The researchers found that the cells in the boys' organoids divided more rapidly and made more synapses than those in organoids derived from their unaffected fathers. They also had elevated expression of genes involved in cell growth, neuronal maturation and synapse formation.
In particular, the boys' organoids had a higher level of expression of FOXG1, a gene that has been associated with the autism-related condition Rett syndrome. This overexpression, the researchers found, resulted in overproduction of neurons that produce a signaling chemical called GABA that inhibits neural activity. None of the boys had mutations in FOXG1, but they did have variations in nearby DNA segments, highlighting the importance of understanding autism-related mutations not just in genes but also in regulatory regions of the genome.
Restoring the activity of the gene responsible for Angelman syndrome can greatly reduce symptoms, a new study
suggests, especially if the gene is restored early
in development.
Angelman syndrome, a condition related to autism that is characterized by a happy disposition, epilepsy, intellectual disability and anxiety and motor deficits, occurs when there is a mutation disabling the maternal copy of the gene UBE3A. In previous mouse model studies, researchers activated the ordinarily silent paternal copy of UBE3A, but found that the mice still displayed repetitive behaviors and anxiety.
The new study suggests, however, that activating UBE3A may prevent some Angelman syndrome symptoms if it is done early enough. A team led by SFARI Investigator Ype Elgersma of Erasmus Medical Center in Rotterdam, the Netherlands, studied mice whose maternal copy of UBE3A had been deactivated. The researchers then turned the gene back on at various stages of development.
They reported in the May 2015 issue of the Journal of Clinical Investigation that when they turned the gene on during embryonic development or on
the first day of life, the mice did not go on to develop anxiety or motor deficits. By contrast, mice that were treated during toddlerhood escaped only
motor deficits, and by adolescence this window had closed as well. Even mice that were treated as adults, however, regained some brain plasticity. The
results suggest that early treatment is key, but that treatment at any age may confer
some benefits.
About 10 percent of autism in boys may result in part from rare genetic mutations inherited from their mothers, according to a study published in the June 2015 issue of Nature Genetics. SFARI Investigators Evan Eichler and Raphael Bernier of the University of Washington and their colleagues examined the exomes — the protein-coding regions of the genome — of 2,377 families from the Simons Simplex Collection, a repository of data from families with one child with autism and unaffected parents and siblings.
The researchers found that even though the mothers in the study did not have autism, they were more likely to have passed down rare mutations to their children with autism than to their unaffected children, at least when it came to 'conserved' genes, ones that are seldom mutated in the general population. The findings are consistent with the idea that women are somehow protected from some of the genetic causes of autism.
On average, the children with the most severe cases of autism had the most of these rare inherited mutations. The researchers estimate that inherited single-letter mutations in DNA contribute to about 7 percent of autism cases; another 3 percent of cases, they propose, result from small copy number variations — regions of DNA that are duplicated or deleted — that contain at least one conserved gene.
Over the past decade, some brain imaging studies of individuals with autism have suggested that their brains are less connected than those of controls, whereas other studies have suggested the opposite. A new study by SFARI Investigator Marlene Behrmann of Carnegie Mellon University in Pittsburgh, Pa., and two co-authors suggests that both findings are correct, and that they are manifestations of a previously undiscovered characteristic of autism spectrum disorders: distorted brain connectivity patterns, often with strong connections in regions where control brains have weak connections, and vice versa.
Behrmann's team, which published its findings in the February 2015 issue of Nature Neuroscience, analyzed resting-state, functional magnetic resonance imaging scans from 73 controls and 68 adults with high-functioning autism from the open-access Autism Brain Imaging Data Exchange.
The brain connectivity patterns of the controls adhered fairly closely to a canonical template, yet those of the individuals with autism varied widely, the researchers found, in terms of both intra- and interhemispheric conductivity, observations consistent with the heterogeneity of the disorder. The researchers found that the individuals with the most idiosyncratic connectivity patterns in certain interhemispheric connections had the most severe autism symptoms. These individual alterations in brain organization may be a core characteristic of high-functioning autism, the team proposes.
Researchers have identified a molecular switch that, when disrupted, locks the UBE3A protein — associated with both autism and Angelman syndrome — in a hyperactive state. Mice with overactive UBE3A, the team found, have an unusually high density of dendritic spines, neuronal protrusions that receive signals from synapses.
The researchers, led by Mark Zylka and SFARI Investigator Ben Philpot of the University of North Carolina at Chapel Hill, began by examining mutations in individuals with Angelman syndrome, who typically have underactive UBE3A. They discovered mutations clustered around a UBE3A region regulated by an enzyme called protein kinase A, which can turn the protein off. In 2014, a whole-exome analysis of the Simons Simplex Collection identified a child with autism who has hyperactive UBE3A and a mutation in precisely this region.
The researchers, who published their findings in the August 13, 2015, issue of Cell, found that in mice, the inactive form of UBE3A is most prevalent at birth and then tapers off over the first week of life, suggesting that mutations that make UBE3A hyperactive may disrupt brain development most during this early stage of development. The study hints at a possible role for UBE3A-inactivating drugs such as Rolipram in treating individuals with autism who have UBE3A mutations.
A new study has linked sound-processing impairment to a copy number variant that is one of the most common causes of autism. Children with deletions in the chromosomal region 16p11.2 process sound markedly more slowly than controls do, researchers reported on February 11, 2015, in Cerebral Cortex.
The researchers, led by SFARI Investigator Timothy Roberts of the Children's Hospital of Philadelphia, used magneto-encephalography (MEG) to study
how quickly the brain responds to sounds in 51 children with 16p11.2 variants who are part of the Simons Variation in Individuals Project, and 45 controls.
They found that children carrying a 16p11.2 deletion produced a particular magnetic response to sound known as M100 about 23 percent later than controls
did. Children with a 16p11.2 duplication produced the M100 signal slightly faster than controls did, though this difference was not statistically
significant. Less than 20 percent of individuals with 16p11.2 variants have autism, but many of the others show language impairments or developmental
delays. The sound-processing delays among children with 16p11.2 deletions occurred across the board, and happened in response to simple stimuli. These
delays may have an even greater impact when it comes to processing more complex sounds such as spoken language, which requires rapid,
ongoing processing.